Computational methods for predicting the effects of genetic variants are used by clinicians for interpreting genetic test results and by researchers for scientific exploration. However, their utility is limited by several issues. First, hundreds of methods are available without clear standards for selection. Additionally, discrepancies among methods regarding the impact of specific variants; challenges in interpreting numerical or categorical predictions (“deleterious”, “pathogenic”, “likely pathogenic” etc.); and the details about the data and logic used for predictions aren’t clear, making it hard to understand results. Furthermore, popular “meta-predictors” designed to combine results from many prediction tools and new AI-based methods have made these transparency and interpretability issues even worse. Users of meta-predictors often don’t know which core predictors matter the most, or how the core predictors redundantly utilize the same features. AI-based predictions hide their processing, taking raw data through a black box and producing outputs without explanation.
This year OpenCRAVAT and CIViC will collaborate in a joint effort to address these issues. As a first step, we will be disseminating pioneering work led by Drs. Vikas Pejaver, Pedja Radivojac, and Steven Brenner, to calibrate the scores of variant effect predictors to strengths of evidence within the ACMG/AMP framework for clinical variant classification. A detailed description of the calibration algorithm can be found in Pejaver et al. 2022.
Clinical geneticists are tasked with interpreting inherited variants identified in genetic testing. This is done with protocols set by professional organizations such as the American College of Medical Genetics (ACMG), the Association for Molecular Pathology (AMP) and others. The ACMG/AMP protocol published in Richards et al. in 2018 classifies variants as Pathogenic or Benign, according to a series of criteria that include molecular properties, family history, population frequencies, functional assays, and computational variant effect predictors, which are the topic of this blog. Each criteria is assessed by rules that measure its “strength of evidence”. In the 2018 guidelines, computational variant effect predictors were not given much weight, because of concerns about their accuracy, interpretability, and transparency. Guidelines for usage of computational tools suggested looking for consensus among multiple variant effect predictors. Many of us disagree with this approach, and we have worked to address these concerns and provide updated recommendations. The work presented in Pejaver et al. AJHG 2022 centers on two criteria known as “BP4” and “PP4”.
The criteria BP4 and PP3 for computational variant effect predictors are included in the RIchards classification framework as supporting criteria for benignity and pathogenicity, respectively:


Table 4 in Pejaver et al. summarizes recommendations for changes in these criteria:

Pejaver et al. suggest that to map variant effect predictor scores onto ACMG/AMP evidence strengths for PP3 and BP4, the first step is to transform the raw scores from a predictor into posterior probabilities of pathogenicity or benignity. The next step is to transform posterior probabilities into positive likelihood ratios, which are more intuitively understood. Finally, raw scores from a variant predictor can be mapped onto posterior probabilities of pathogenicity or benignity. All mathematical details are provided in Pejaver et al.
Over the first half of 2025, we hope to make these mappings for all variant effect predictors easily available in the OpenCRAVAT store and to disseminate them on CIViC. You can find the following calibrated variant effect predictors in the OpenCRAVAT store as of 12/9/2024. The store is available both through run.opencravat.org and your local installation of OpenCRAVAT. More calibrated variant effect predictors will be coming soon!
| Predictor Name | Methodology | Citation |
| BayesDel | Meta-score to assess deleteriousness | Feng, Bing-Jian. “PERCH: A Unified Framework for Disease Gene Prioritization.” Human mutation vol. 38,3 (2017): 243-251. |
| CADD | convolutional neural network | Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res.2018 Oct 29. |
| CADD exome | convolutional neural network | Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res.2018 Oct 29. |
| FATHMM | Hidden Markov models | Shihab, H. A., Gough, J., Mort, M., Cooper, D. N., Day, I. N., & Gaunt, T. R. (2014). Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Human genomics, 8(1), 11. |
| GERP++ | Nucleotide conservation | Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., & Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS computational biology, 6(12), |
| PHYLOP | Nucleotide conservation | Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., & Siepel, A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome research, 20(1), 110-121. |
| REVEL | Meta-predictor, Random Forest | Ioannidis, N. M., Rothstein, J. H., Pejaver, V., et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. The American Journal of Human Genetics, 99(4), 877-885. |
| SIFT | Multiple sequence alignment | Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC (2016) SIFT missense predictions for genomes. Nat Protocols 11: 1-9. |
| VEST4 | Random forest | Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013;14 Suppl 3(Suppl 3):S3. |
| PrimateAI | Deep residual neural network | Sundaram, L., Gao, H., Padigepati, S.R. et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50, 1161–1170 (2018). |
When you run an annotation job with a calibrated variant effect predictor, you will see two new columns: ACMG/AMP Benign (BP4) and ACMG/AMP Pathogenic (PP3).
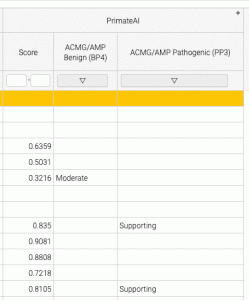
The mapping to clinical evidence strength may be different from the threshold for pathogenicity (or deleteriousness, damage etc) For example, in the results shown below, the PrimateAI raw score of 0.835 is above the recommended threshold for a damaging variant, but with respect to ACMG/AMP clinical Pathogenicity, the evidence that the variant is pathogenic is only “supporting” (vs. very strong, strong, or moderate).
BP4 and PP3 strength of evidence can also be visualized in the widget for each variant effect predictor, such as this one for REVEL.

Please let us know your thoughts and experience with this new feature at https://github.com/KarchinLab/open-cravat/discussions or by emailing us at support@opencravat.org